Ved fødslen fødes mennesket med en umoden kranium dannet af knogler, som endnu ikke har nået deres totale lukning. Denne anatomiske tilstand gør det muligt for hjernen at vokse og modnes med alderen.
Men i sjældne tilfælde svejsning af disse knogler Er gjort for tidligt og denne anomali påvirker både hjernens morfologi og ansigtet, hvilket giver et udseende af misdannelse meget karakteristisk. Det er det, der definerer Crouzons syndrom, en patologi sjælden genetik som påvirker cirka 1/50000 fødsler.
Gå direkte til det afsnit, der interesserer dig
Hvad er Crouzon syndrom?
Navnet på Crouzon syndrom kommer fra den franske neurolog Louis Edouard Crouzon, som beskrev det for første gang i begyndelsen af det 20. århundrede.
Dette er en medfødt misdannelse (til stede ved fødslen), af genetisk oprindelse, knyttet til en mutation af FGFR2-genet placeret på kromosom 10. På det anatomopatologiske plan er denne anomali karakteriseret ved en for tidlig suturering af kraniets knogler (kraniosynostose), hvilket forårsager atypisk vækst af hjernen og de organer, der udgør ansigtet.
Barnet, der bærer dette syndrom, kan vise mere eller mindre alvorlige neurologiske og funktionelle komplikationer, når det vokser op. Disse komplikationer kan dog forebygges eller reduceres væsentligt, så længe sygdommen diagnosticeres og behandles i tide.
Fra et epidemiologisk synspunkt er Crouzon syndrom den mest almindelige sjældne genetiske abnormitet, der påvirker kraniets knogler.
Hvordan genkender man Crouzon syndrom?
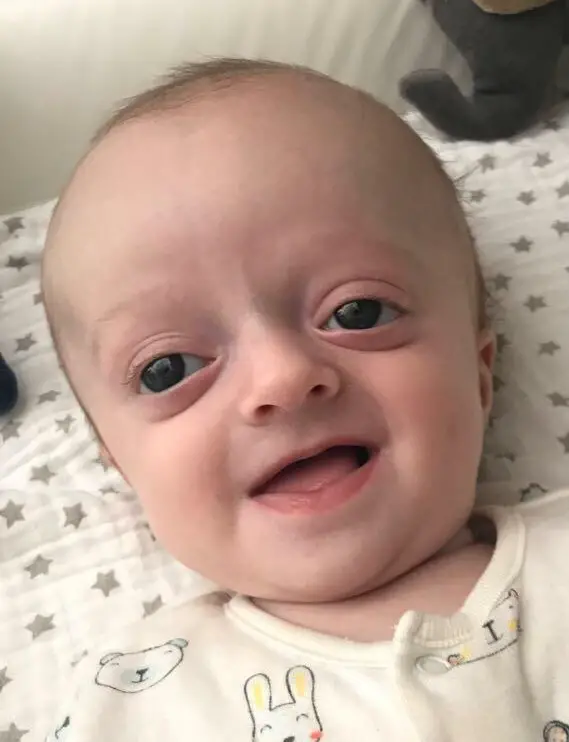
Det er indlysende, at Crouzon syndrom er identificeret ved fødslen, men de fleste af de symptomer, der definerer dette syndrom, er tydelige i løbet af det første leveår.
Generelt præsenterer spædbørn, der er berørt, et billede i det væsentlige benet, som oftest kommer til udtryk ved:
A brachycefali: hvilket svarer til en forstørrelse af kraniet på parietalniveau og en udfladning af baghovedet, hvilket giver indtryk af, at baghovedet er højere end panden.
A proptose: begreb, der betegner en udgravning af øjnene, som er et resultat af den for tidlige forbening af knoglerne, der udgør øjenæblerne. Dette fremspring kan fremkalde synsproblemer hos barnet.
En horbital hypertelorisme ou ydem kasseret : symptom karakteriseret ved en unormal stigning i afstanden mellem de to øjne. Denne misdannelse observeres i mange genetiske sygdomme, såsom trisomi 18, Apert syndromOsv
Un næbformet næse : hvis det ikke behandles kirurgisk, kan dette symptom være ansvarlig for åndedrætsbesvær, lige fra simpelt åndedrætsbesvær til obstruktivt søvnapnøsyndrom.
A pprojektion af underkæben (underkæben) fremad, hvilket er mere tydeligt, når det parres med en overkæbehypoplasi. Dette ændrer mundhulens anatomi og påvirker nogle gange tygning og fonation.
Derudover kan andre mindre almindelige symptomer og komplikationer forekomme inkonsekvent hos børn med Crouzon. Blandt disse tegn er:
Des høreproblemer: 55 % af børn med Crouzon syndrom har en stærkt reduceret akustisk kapacitet på grund af, at de er født uden øregange.
Des fælles problemer : på cervikal niveau, rapporteret hos 30 % af patienterne.
Hydrocephalie et Arnold-Chiari misdannelse : disse er to komplikationer, der opstår på lang sigt, og deres tilstedeværelse retfærdiggør generelt fraværet af tilstrækkelig håndtering.
Acanthosis Nigricans: denne dermatologiske tilstand, som oftest er en markør for insulinresistens hos børn, kan også indgå i Crouzons syndrom, hvor den er karakteriseret ved tykkere og mørkere hud.
Hvad er årsagerne til denne tilstand?
Som nævnt i definitionen er Crouzon syndrom en genetisk sygdom, hvis oprindelse ligger på "FGFR2" gen hvor der er "fejl" eller "mutation".
Dette gen er involveret i at kontrollere produktionen af proteiner, der er ansvarlige for knogleudvikling og vækst. Dens mutation vil derfor forårsage en acceleration af ossifikationsprocessen af hele eller dele af kraniale suturer.
Overførselsmåden af sygdommen er autosomal dominant. Det vil sige, at en person, der bærer mutationen, har 50 % risiko for at overføre den til deres fremtidige børn.
Hvad med prognosen?
Du skal vide, at denne sygdom, selvom det er uhelbredelig, er generelt altid god prognose ! Et barn med dette syndrom kan leve et næsten normalt liv, forudsat at dets sygdom diagnosticeres så tidligt som muligt og operationen udføres i tide.
Derudover, hvis det kan være betryggende, bortset fra de forskellige komplikationer, som vi allerede har nævnt ovenfor, har et barn med dette syndrom intellektuelle kapaciteter, der ligner dem hos et sundt individ. Det kognitive evner et intellektuelle er ikke berørt.
Behandling af Crouzon syndrom
Selvom der er flere undersøgelser og undersøgelser, der er blevet udført med det formål at genetisk helbrede Crouzon syndrom, har ingen resulteret i forsøg på mennesker.
Behandlingen af denne sygdom er derfor i det væsentlige symptomatisk, og sigter mod forhindre dets komplikationer og dermed forbedre livskvaliteten for det berørte barn.
For en bedre prognose for sygdommen skal barnet have gavn af en tværfaglig pleje, der involverer fagpersoner fra forskellige specialer, såsom praktiserende læger, børnelæger, kraniofaciale kirurger, talepædagoger, psykologer eller børnepsykiatere mv.
De vigtigste terapier er:
kirurgi
De fleste børn med dette syndrom er kandidater til operation, fordi det repræsenterer et af de vigtigste trin i deres pleje.
Det giver dem mulighed for at rette deres kraniofacial dysmorfi og dermed vil give deres hjerner chancen for at udvikle sig så normalt og med så få komplikationer som muligt.
Et af disse indgreb er kranioplastik som kirurger bruger til at behandle eller forebygge en evt hydrocephalus, og også lette skiltene tilintrakraniel hypertension (kvalme, opkastning, hovedpine osv.).
På samme måde ser operation af næsepassager og mundhule ud til at være lige så vigtig for at forhindre åndedrætsproblemer såsom søvnapnø.
Exophthalmos kan også behandles kirurgisk, dette vil ikke kun forbedre den æstetiske side af barnet, men vil også minimere risikoen for udtørring og infektion i øjnene.
Kæbeindgreb for at forbedre underkæbens fremspring og deformiteten af overkæben.
I lyset af store luftvejsproblemer, en tracheotomi kan gøres omgående.
Remarque : det skal bemærkes, at det bedste tidspunkt at ty til kirurgi (kranioplastik) er de første 12 måneder af livet, da knoglerne i denne alder lettere modelleres og tilpasser sig bedre til kirurgiske korrektioner. Ikke desto mindre er risikoen for gentagelse af suturerne så meget desto højere, som barnets alder er lav.
Taleterapi
Logopædens rolle består i at minimere de komplikationer, der er forbundet med misdannelser af det fonatoriske apparat. Det kan hjælpe barnet til at sluge sikkert, lære dem at forbedre deres sprog og vejrtrækning. I nogle tilfælde, hvor der er en forbundet kognitiv svækkelse, kan talepædagogen vælge den passende terapi.
ergoterapi
Denne terapeutiske tilgang beskæftiger sig primært med problemer med at spise, klæde sig på, lære at skrive, følelsesmæssig håndtering samt anden personlig pleje relateret til dagligdags aktiviteter.
Psykoterapi
Et barn, der er diagnosticeret med en sjælden patologi som Crouzons syndrom, risikerer at støde på socioaffektive og følelsesmæssige problemer i barndommen eller senere i voksenalderen, som kun vil fremhæve hans lidelse. Det er derfor vigtigt at fokusere på disse patienters, men også deres forældres mentale sundhed og velbefindende, for at hjælpe dem bedre med at tage sig af deres børn med Crouzon syndrom.
Var denne artikel nyttig for dig?
Angiv din påskønnelse af artiklen
Læsernes vurdering 3.7 / 5. Antal stemmer 3
Hvis du har haft gavn af denne artikel
Del det gerne med dine kære
Tak for dit afkast
Hvordan kan vi forbedre artiklen?
Anas Boukas( Fysioterapeut, fysioterapeut, konsulent )
Jeg hedder Anas Boukas og er fysioterapeut. Min mission? At hjælpe mennesker, der lider, før deres smerter forværres og bliver kroniske. Jeg tror også på, at en uddannet patient i høj grad øger deres chancer for helbredelse. Det er derfor, jeg har skabt Healthforall Group, et netværk af medicinske websteder, i samarbejde med flere sundhedsprofessionelle.

")



")
")
")
